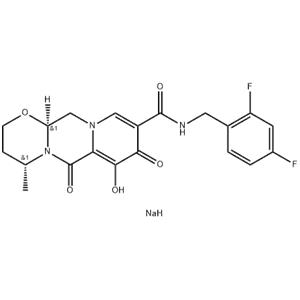
| Description |
Dolutegravir sodium is an inhibitor of HIV-1 integrase-catalyzed strand transfer with IC50 of 2.7 nM. |
| Related Catalog |
Signaling Pathways >>
Metabolic Enzyme/Protease >>
HIV Integrase
Signaling Pathways >>
Anti-infection >>
HIV
Research Areas >>
Infection
|
| Target |
IC50: 2.7 nM (HIV-1 integrase)[1]
|
| In Vitro |
The EC50 of Dolutegravir (S/GSK1349572) against HIV-1 is 0.51 nM
in PBMCs, 0.71 nM in MT-4 cells, and 2.2 nM in the PHIV assay, which
uses a pseudotyped self-inactivating virus. The 50% cytotoxic
concentrations (CC50) for Dolutegravir in proliferating IM-9, U-937,
MT-4, and Molt-4 cells are 4.8, 7.0, 14, and 15 μM, respectively. In
unstimulated and stimulated PBMCs, the CC50 are 189 μM and 52 μM,
respectively. Based on the EC50 of Dolutegravir against HIV-1 in PBMCs
(i.e., 0.51 nM), this translates to a cell-based therapeutic index of at
least 9,400[1].
|
| In Vivo |
Following a single intravenous (IV) administration of
Dolutegravir, the plasma clearance is low in rats (0.23 mL/min/kg) and
monkeys (2.12 mL/min/kg). The half-lives in the rat and monkey are
similar, approximately 6 h, and the steady-state volume of distribution
(VSS) is low. Following oral administration, Dolutegravir is rapidly
absorbed with a high oral bioavailability when administered as a
solution to fasted male rats and a single monkey (75.6 and 87.0%,
respectively). Dolutegravir exposure (Cmax and AUC) increased with
increasing dose following oral administration of a suspension to
non-fasted rats up to 250 mg/kg and non-fasted monkeys up to 50 mg/kg,
although the increase is less than proportional[2].
|
| Cell Assay |
In vitro growth inhibition (cytotoxicity) studies are conducted
with S/GSK1349572 (0.16, 0.8, 4, and 20 nM) in proliferating human
leukemic and lymphomic cell lines (IM-9, U-937, MT-4, and Molt-4) as
well as in stimulated and unstimulated human PBMCs. ATP levels are
quantified by using the CellTiter-Glo luciferase reagent to measure the
ability of a compound to inhibit cell growth as an indicator of the
compound's potential for cytotoxicity[1].
|
| Animal Admin |
For rat and monkey PK studies, Dolutegravir is administered as the
free acid or the sodium salt. All doses are presented in terms of the
free acid. Dolutegravir is administered by intravenous (IV) short-term
(within 2 min) bolus (1 mg/kg) to three male rats and two male monkeys.
For single oral administration, Dolutegravir as a solution (5 mg/kg) is
administered to three fasted male rats and two fasted male monkeys.
Dolutegravir is administered as single oral doses of 5, 50, 100, and 250
mg/kg to non-fasted male rats (n=2/dose level) and 3, 10, and 50 mg/kg
to non-fasted female monkeys. For intravenous administration, blood
samples are collected from rats (0.2 mL via jugular vein cannula) and
monkeys (approximately 0.2 or 0.5 mL via saphenous vein in a hindlimb)
into Na2EDTA-treated syringes at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24
h. For oral administration, samples are collected at 0.25 (rats only),
0.5, 1, 2, 4, 6 [rats (solution and suspension) and monkey (solution
only)], 8, and 24 h. Following collection, the blood is immediately put
on wet ice and then centrifuged within an hour at 1740 g for 10 min at
4°C to obtain plasma. All samples are stored at approximately -20°C or
colder prior to analysis by using a method based on protein
precipitation and LC-MS/MS analysis.
|
| References |
[1]. Kobayashi
M, et al. In Vitro antiretroviral properties of S/GSK1349572, a
next-generation HIV integrase inhibitor. Antimicrob Agents Chemother.
2011 Feb;55(2):813-21. [2]. Moss
L, et al. The comparative disposition and metabolism of dolutegravir, a
potent HIV-1 integrase inhibitor, in mice, rats, and monkeys.
Xenobiotica. 2015 Jan;45(1):60-70.
|

 China
China