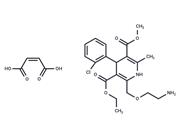
| Name | AM095 |
| Description | AM095 is a potent LPA1 receptor antagonist with IC50 values of 0.98 and 0.73 μM for recombinant human or mouse LPA1 respectively. |
| Kinase Assay | Known amounts of AM095 (diluted in DMSO) or vehicle (DMSO) are added to 25 to 40 μg of hLPA1/CHO or mLPA1/CHO membranes and 0.1 nM [35S]-GTPγS in buffer (50 mM HEPES, 0.1 mM NaCl, 10 mM MgCl2, 50 μg/mL saponin, pH 7.5) containing 0.2% fatty acid-free human serum albumin and 5 μM GDP. To test for LPA1 antagonist activity, the ability of AM095 to inhibit GTPγS binding stimulated by 900 nM LPA (18:1) is measured. Alternatively, to test for agonist effects, the ability of AM095 to stimulate GTPγS binding in the absence of LPA is measured. Reactions are incubated for 30 min at 30°C, before harvesting membranes onto glass filter binding plates (UniFilter GF/B) and washing three times with cold buffer containing 50 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2 using a Brandel 96-tip cell harvester. Plates are dried and then cpm are evaluated by using a Packard TopCount NXT microplate scintillation counter[1]. |
| In vitro | AM095 is a potent LPA1 receptor antagonist because it inhibits GTPγS binding to Chinese hamster ovary (CHO) cell membranes overexpressing recombinant human or mouse LPA1 with IC50 values of 0.98 and 0.73 μM, respectively. AM095 inhibits LPA-driven chemotaxis of CHO cells overexpressing mouse LPA1 (IC50=778 nM) and human A2058 melanoma cells (IC50=233 nM). The IC50 of AM095 in the human LPA1 GTPγS binding assay is comparable with that of our previously published compound AM966 (IC50=0.98±0.17 μM) and the Debio-0719 compound (IC50=0.60±0.04 μM)[1]. AM095 inhibits the LPA-induced calcium flux of CHO cells stably transfected with human or mouse LPA1. The IC50 for AM095 antagonism of LPA-induced calcium flux of human or mouse LPA1-transfected CHO cells is 0.025 and 0.023 μM, respectively[2]. |
| In vivo | AM095 exhibits high oral bioavailability and moderate half-life, demonstrating tolerability in both rats and dogs following oral and intravenous administration. In rats, an oral dose (10 mg/kg) of AM095 results in peak plasma concentrations (Cmax) of 41 μM at 2 hours, decreasing to 10 nM by 24 hours. Conversely, an intravenous dose (2 mg/kg) leads to a Cmax of 12 μM within 15 minutes, similarly diminishing to approximately 10 nM by 24 hours, with a half-life (t1/2) of 1.79 hours. In dogs, an oral administration of 5 mg/kg achieves a peak plasma concentration of 21 μM within 15 minutes, falling to 10 nM by 24 hours, whereas an intravenous dosage (2 mg/kg) yields a Cmax of 11 μM in 15 minutes, reducing to 15 nM by 8 hours, and a t1/2 of 1.5 hours[1]. |
| Storage | Powder: -20°C for 3 years | In solvent: -80°C for 1 year | Shipping with blue ice. |
| Solubility Information | DMSO : 5.5 mg/mL (11.49 mM), Sonication is recommended.
|
| Keywords | LPL Receptor | AM 095 | inhibit | Lysophospholipid Receptor | Inhibitor | AM-095 | AM095 |
| Inhibitors Related | Radioprotectin-1 | Fingolimod hydrochloride | SLP9101555 | Tyloxapol | ASP-4058 | Ki16198 | CYM50260 | Tetradecyl Phosphonate | Siponimod | Fingolimod | S1PR1 modulator 1 | Ozanimod |
| Related Compound Libraries | Bioactive Compound Library | Membrane Protein-targeted Compound Library | Anti-Obesity Compound Library | Inhibitor Library | NO PAINS Compound Library | Anti-Fibrosis Compound Library | Bioactive Compounds Library Max | Preclinical Compound Library | GPCR Compound Library | Anti-Cancer Active Compound Library |

 United States
United States