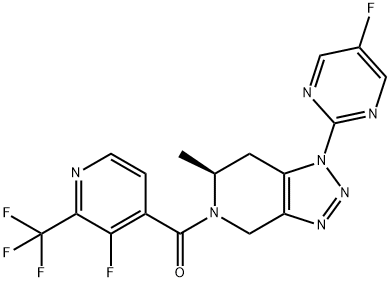
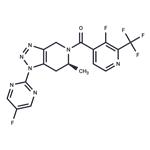
JNJ-55308942 is a high-affinity, selective, brain-penetrant P2X7 functional antagonist (hP2X7: IC50=10 nM, Ki=7.1 nM; rP2X7: IC50=15 nM, Ki=2.9 nM). JNJ-55308942 is orally bioavailable, binds to brain P2X7 and blocks IL-1β release from adult rodent brain[1][2].
JNJ-55308942 shows pKis of 8.1and 8.5 for recombinant human and rat P2X7 channels, respectively. In human blood and in mouse blood and microglia, JNJ-55308942 attenuates IL-1β release in a potent and concentration-dependent manner[2].
JNJ-55308942 (30 mg/kg; p.o.) attenuates LPS-induced microglial activation in mice[2].In a model of Bacillus Calmette-Guerin (BCG)-induced depression, JNJ-55308942 dosed orally (30 mg/kg), reversed the BCG-induced deficits of sucrose preference and social interaction. After oral dosing, the compound exhibited both dose and concentration-dependent occupancy of rat brain P2X7 with an ED50 of 0.07 mg/kg. The P2X7 antagonist (3 mg/kg, oral) blocked Bz-ATP-induced brain IL-1β release in conscious rats, demonstrating functional effects of target engagement in the brain[2]. JNJ-55308942 (5 mg/kg; p.o.) shows the F, Vss, CL, Cmax and AUC24h values are 81%, 1.7 L/kg, 3.7 mL min/kg, 1747 ng/mL, and 17549 (ng/mL) h, respectively[1].
[1]. Bhattacharya A, et al. Neuropsychopharmacology of JNJ-55308942: evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacology. 2018;43(13):2586-2596. [2]. Chrovian CC, et al. A Dipolar Cycloaddition Reaction To Access 6-Methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines Enables the Discovery Synthesis and Preclinical Profiling of a P2X7 Antagonist Clinical Candidate. J Med Chem. 2018;61(1):207-223.