![N-[(1S,5R)-7,9-dimethyl-7,9-diazabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide dihydrochloride Structure](/CAS/GIF/160472-97-9.gif)
N-[(1S,5R)-7,9-dimethyl-7,9-diazabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide dihydrochloride
- Product NameN-[(1S,5R)-7,9-dimethyl-7,9-diazabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide dihydrochloride
- CAS160472-97-9
- MFC17H24ClN5O
- MW349.86
- EINECS
- MOL File160472-97-9.mol
Usage And Synthesis
Concomitant with the emesis provoked by radiation and cytotoxic drugs is an
increase in 5-hydroxytryptamine (5-HT) concentration in the brain stem and intestinal
mucosa, which in turn stimulates the 5-HT3 receptors on the adjacent vagal
afferent nerves. The depolarization of the vagal afferents is responsible for inducing the vomiting reflex. Indisetron, a 5-HT3/5-HT4 antagonist, has, therefore, been
launched in Japan as an anti-emetic. Since 5-HT4 is implicated in intestinal motility,
dual antagonism should improve the anti-emetic effect, although this remains to be
demonstrated. The diazabicycloamine portion of indisetron is prepared in four steps
starting with methylamine and bromoacetaldehyde dimethylacetal. Coupling to the
indazole core is accomplished via the acid chloride of 1H-indazole-3-carboxylic acid.
In vitro, indisetron displaced [3H]GR-65630 binding to the 5-HT3 receptor in rat brain
membranes in a concentration-dependent manner with a pKi value of 8.77, which is
comparable to the activities of other 5-HT3 antagonists such as granisetron and ondansetron.
In animal models (ferrets and dogs), indisetron reduced the number and
duration of cisplatin-induced emetic episodes when administered orally at 0.1–1mg/kg
prior to cisplatin treatment. Compared to granisetron and ondansetron, there were no
significant differences in the frequency of vomiting; however, the duration was significantly
reduced with indisetron. After the administration of anticancer agents, including
cisplatin, in phase II and phase III clinical trials, indisetron prevented vomiting
in 62%of patients (67 of 108) for 24 h. It faired even better in a phase III clinical study
where the anticancer agents did not include cisplatin; emesis was averted in 34 of 40
patients (85%). In a single oral dose study (4, 8, 16, and 32mg under fasting conditions
or a 16mg dose post-prandial) in healthy males, indisetron showed high oral
bioavailability (nearly 100%), and Cmax and AUC increased in a dose-dependent
manner. While four subjects experienced adverse effects, they were considered mild
and not clinically significant. In a phase I study comprised of 16mg of indisetron
administered b.i.d. over 7 consecutive days, the adverse reactions were also deemed
clinically insignificant although it was noted that two patients presented with constipation
and an increase in serum amylase. Steady state plasma levels were achieved
by day 3. Repeated administration did not change the pharmacokinetics
and accumulation of indisetron. Indisetron is metabolized by a host of CYP enzymes
(1A1, 2C9, 2D6, and 3A4) in the liver; however, it is unlikely to cause drug interactions
at clinical doses because the plasma concentrations are lower than those necessary for
CYP inhibition. Finally, as a 5-HT3 antagonist, indisetron should avoid the adverse
effects of the current dopamine-blocking anti-emetics, such as, sedation, ataxia, diarrhea,
and tasikinesia, making it a viable alternative to existing therapy.
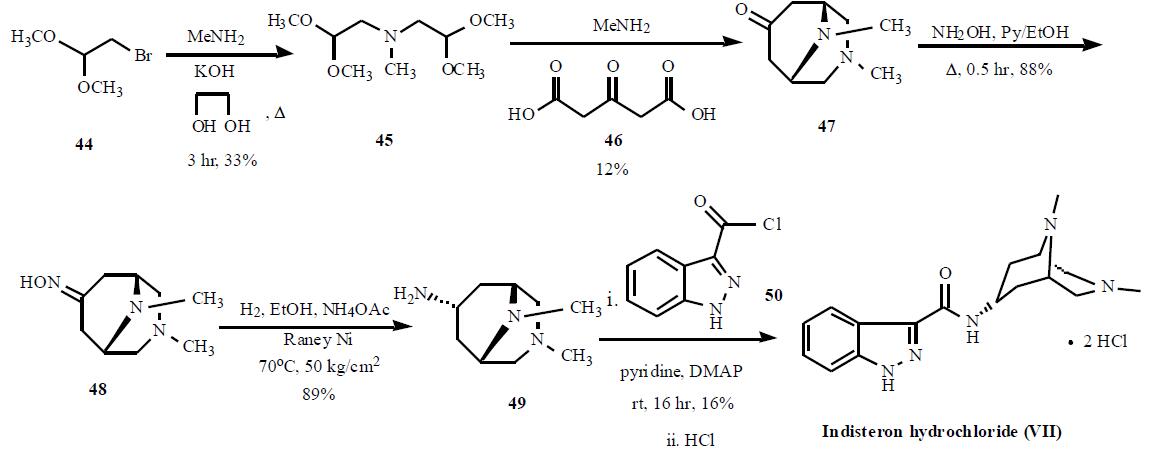
The synthesis is highlighted in the Scheme.
Bromoacetaldehyde dimethyl acetal (44) was condensed with
methylamine with KOH in refluxing ethyleneglycol for 3 hr
to give 33% yield of bis(2,2-dimethoxyethyl)amine (45),
which was cyclized with acetonedicarboxylic acid (46) and
methylamine to generate 3,9-dimethyl-3,9-diazabicyclo-
[3.3.1]nonan-7-one (47) in 12% yield. Compound 47 was
reacted with hydroxylamine in refluxing pyridine and ethanol
mixture to give corresponding oxime 48 in 88% yield,
which was subsequently reduced with hydrogen over Raney
Ni in hot ethanol in the presence of ammonium acetate at 50
kg/cm2 to give amine 49 in 89% yield. Compound 49 was condensed with 1H-indazole-3-carbonyl chloride (50) in
pyridine with catalytic amount of DMAP to give crude
indisteron free base, which was re-crystallized from
chloroform/hexane to give indisteron free base as colorless
crystals in 16% yield. Finally, the free base was treated with
hydrogen chloride to give indisteron hydrochloride (VII).
N-[(1S,5R)-7,9-dimethyl-7,9-diazabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide dihydrochloride Supplier
N-[(1S,5R)-7,9-dimethyl-7,9-diazabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide dihydrochloride manufacturers

PROMPT×
PROMPT
The What'sApp is temporarily not supported in mainland China
The What'sApp is temporarily not supported in mainland China
Cancel
Determine