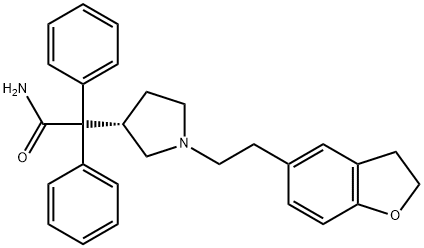
Darifenacin is a novel muscarinic M3 selective antagonist for the once-daily oral
treatment of urinary incontinence and overactive bladder. The majority of overactive
bladder symptoms are thought to result from the overactivity of the detrusor
muscle, which is primarily mediated by acetylcholine-induced stimulation of muscarinic
M3 receptors in the bladder. Consequently, antimuscarinic agents have become
the mainstay of overactive bladder treatment. Darifenacin has a higher level
of M3 selectivity than the previously marketed antimuscarinic agents. It has Ki
values of 16nM for M1, 50 nM for M2, and 1.6 nM for M3 receptors. It is slightly
more M3 selective than solifenacin (M1:Ki=25 nM, M2:Ki=126 nM,
M3:Ki=10 nM), which was launched in 2004. Darifenacin is significantly more
selective than other muscarinics such as tolterodine, oxybutynin, and trospium,
which are all essentially equipotent against M1, M2, and M3 receptors. In addition,darifenacin demonstrates greater effect on tissues in which the predominant receptor
type is M3 rather than M1 or M2. In vitro darifenacin inhibits carbacholinduced
contractions with greater potency in isolated guinea-pig bladder (M3) than
in guinea-pig atria (M2) or dog saphenous vein (M1). In animal models, it shows
greater selectivity for inhibition of detrusor contraction over salivation or tachycardia.Darifenacin is supplied as a controlled
release formulation, and the recommended dosage is 7.5 mg once, daily.
Darifenacin is rapidly and completely absorbed from the GI tract after oral administration,
with maximum plasma levels achieved after about 7 h. The elimination
half-life is approximately 3 h, but because of the controlled release characteristics of
the formulation, the drug is suitable for once-daily dosing. Steady-state plasma
levels are achieved within 6 days of commencing treatment. Darifenacin exhibits
high-protein binding (98%), a volume of distribution of 163 L, and a clearance of
40 L/h. It has low oral bioavailability (15–19%) due to extensive first-pass metabolism
by CYP3A4 and CYP2D6, but this can be saturated after multiple administrations.
The major circulating metabolites are produced by monohydroxylation
and N-dealkylation; however, none contribute significantly to the overall clinical
effect of darifenacin. Approximately 58% of the dose is excreted in urine and 44%
in feces; only a small percentage (3%) of the excreted dose is unchanged darifenacin.