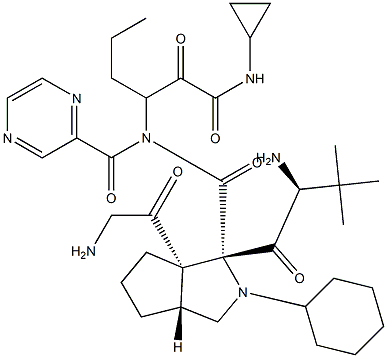
Telaprevir
Bezeichnung:Telaprevir
CAS-Nr402957-28-2
Englisch Name:Telaprevir
CBNumberCB41519837
SummenformelC36H53N7O6
Molgewicht679.85
MOL-Datei402957-28-2.mol
Telaprevir physikalisch-chemischer Eigenschaften
| Schmelzpunkt | 116-123°C |
| Dichte | 1.25 |
| storage temp. | Sealed in dry,Store in freezer, under -20°C |
| Löslichkeit | Chloroform, Methanol (Slightly) |
| pka | 11.84±0.20(Predicted) |
| Aggregatzustand | Solid |
| Farbe | Pale Yellow to Light Yellow |
| S-Sätze: | 24/25 |
| HS Code | 29339900 |
| Giftige Stoffe Daten | 402957-28-2(Hazardous Substances Data) |
Gefahreninformationscode (GHS)
-
Bildanzeige (GHS)

-
Alarmwort
Warnung
-
Sicherheit
P201:Vor Gebrauch besondere Anweisungen einholen.
P202:Vor Gebrauch alle Sicherheitshinweise lesen und verstehen.
P280:Schutzhandschuhe/Schutzkleidung/Augenschutz tragen.
P308+P313:BEI Exposition oder falls betroffen: Ärztlichen Rat einholen/ärztliche Hilfe hinzuziehen.
P405:Unter Verschluss aufbewahren.
P501:Inhalt/Behälter ... (Entsorgungsvorschriften vom Hersteller anzugeben) zuführen.
Telaprevir Chemische Eigenschaften,Einsatz,Produktion Methoden
-
Beschreibung
The hepatitis C virus (HCV) protease inhibitor telaprevir (VX-950, MP- 424,LY-570310) was approved by the U.S. FDA in May 2011 for the treatment of genotype 1 chronic HCV infection in adult patients in combination with peginterferon alfa and ribavirin (PR). Telaprevir and boceprevir (vide supra) are the first two HCV protease inhibitors to be approved for treatment of HCV infection. Telaprevir is a HCV NS3-4A protease inhibitor that exerts its antiviral effect by blocking the release of nonstructural viral proteins from a polyprotein precursor. Telaprevir is a potent inhibitor of the protease (IC50=10 nM) and is active in cell culture (HCV 1b replicon assay, EC50=354 nM). Telaprevir was identified from efforts to truncate a decamer peptide inhibitor derived from the natural substrate NS5A-5B and was guided by structure-based design. The ketoamide group of telaprevir forms a covalent, reversible bond with the active site serine hydroxyl of the protease and compensates for the loss of affinity resulting from truncation of the peptide. Despite the presence of the reactive keto-amide group, telaprevir is >500-fold less potent against other serine proteases. Synthesis of the key octahydrocyclopenta[c]pyrrole-1-carboxylic acid fragment of telaprevir is achieved by a-deprotonation of Boc-protected 3-azabicyclo[3.3.0]nonane followed by reaction with CO2 and resolution of the racemic acid. Alternatively, deprotonation is carried out in the presence of a chiral amine to give the enantiomerically enriched acid. -
Chemische Eigenschaften
Telaprevir is White Solid -
Originator
Eli Lilly (United States) -
Verwenden
Labeled Telaprevir, intended for use as an internal standard for the quantification of Telaprevir by GC- or LC-mass spectrometry. -
Verwenden
A labelled peptidomimetic inhibitor of hepatitis C virus protease. -
Verwenden
Telaprevir is a peptidomimetic inhibitor of hepatitis C virus protease. -
Definition
ChEBI: An oligopeptide consisting of N-(pyrazin-2-ylcarbonyl)cyclohexylalanyl, 3-methylvalyl, octahydrocyclopenta[c]pyrrole-1-carboxy, and 3-amino-N-cyclopropyl-2-oxohexanamide residues joined in sequence. Used f r treatment of chronic hepatitis C virus genotype 1 infection. -
Trademarks
Incivek -
Clinical Use
Telaprevir is a potent peptide mimetic inhibitor of Hepatitis C virus (HCV) and works via covalent reversible binding to the NSV-3A protease enzyme. Telaprevir was discovered and developed by Vertex pharmaceuticals. The drug is marketed as an oral treatment for HCV infection in combination with Peg interferon and ribavarin for patients who are refractory to the initial standard therapy. The initial SAR studies and the discovery of teleprevir have been published. In addition, a full review of the discovery process that led to the development of telaprevir, including several iterations of the syntheses of teleprevir leading to the process route, has been reported. -
Synthese
For preparation of bulk API, a convergent synthetic strategy was utilized as described in the scheme. Retrosynthetically the penultimate intermediate 212, which was coupled with amine 213 for the final step, was prepared by coupling bicyclic amine 217 with amino acids 216 and 215 and then with pyrazine acid 214.
In the early stages of development, the cyclopropyl amide fragment 213 was made using a MCR coupling sequence by reacting aldehyde 218 with cyclopropyl isocyanide (219) and triflouroacetic acid to give amide alcohol 220 in 85% yield. Removal of the Cbz group was accomplished via hydrogenolysis to provide key cyclopropyl amide alcohol 213 in 95% yield. While this route was shorter in terms of steps, it was not amenable to large-scale preparation due to difficulties associated with the handling of isocyanide 219.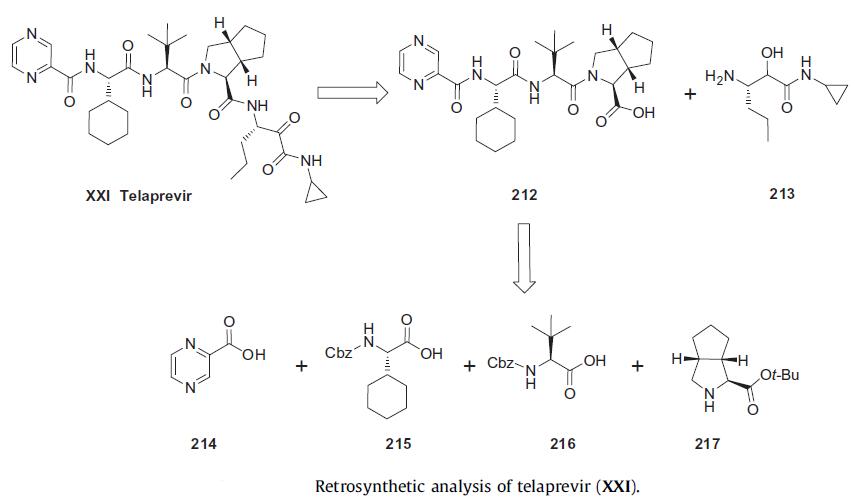
Thus, for large-scale synthesis, the route depicted in Scheme 35 was utilized. Commercially available Cbz-protected amino acid 221 was converted to the corresponding Weinreb amide 222 using CDI as the activating agent. This was followed by LAH reduction to give aldehyde 218 in 73% yield from 221. Aldehyde 218 was reacted with sodium cyanide under neutral to mildly basic conditions allowing for easy workup of the cyanohydrin, which was immediately hydrolyzed by refluxing in 4 N hydrochloric acid in dioxane to deliver hydroxy acid HCl salt 223. Since the formation of the acyloin resulted in removal of the Cbz protecting group, reinstallation of this protecting group preceded conventional amide bond formation through the intermediacy of the succinate ester of 224. This provided the desired amide alcohol 220 in 56% yield from 223. Hydrogenolysis of Cbz carbamate 220 then furnished the requisite intermediate amine (213) in 73% yield.
The large-scale synthesis of bicyclic pyrrolidine 231 was accomplished as described in the following scheme. Commercially available 3-azabicyclo[ 3.3.0]nonane hydrochloride (225) was first protected as the corresponding Boc carbamate 226 in 90% yield. Deprotonation of the bicyclic pyrrolidine carbamate 226 with sec-BuLi and sequential quench with bubbling carbon dioxide gas followed by sodium hydrogensulfate resulted in racemic acid 227 in 80% yield. Racemate 227 was resolved using (S)-tetrahydronapthalamine (228) in ethyl acetate and isopropanol at 70¨C75??C. This mixture was allowed to cool down slowly to effect the crystallization of the optically enriched chiral salt 229 in 83% yield with greater than 99.5% ee. This enatioenriched salt was free based with sodium hydrogen sulfate and converted to t-butyl ester 230 using Boc anhydride and DMAP. The secondary amine of 230 was liberated using methane sulfonic acid at room temperature followed by salt formation with oxalic acid in isopropyl acetate to give oxalic acid salt 231 in 81% yield over 3 steps.
With the synthesis of the key intermediates complete, sequential coupling events were then executed to complete the synthesis of teleprevir (following scheme). Fragment 217 was coupled with the Cbz-protected valine (216) using EDCI and HOBt to give intermediate 232 in 87% yield. Similarly, after removal of the Cbz group of 232 via catalytic hydrogenolysis, the resulting amine was coupled with cyclohexyl amino acid 215 to give dipeptide intermediate 233 in 89% yield over 2 steps. Sequential cleavage of the Cbz group in 233 followed by CDI-mediated coupling with commercially available pyrazine acid 214 gave rise to the expected pyrazine amide intermediate. Subsequent hydrolysis of the t-butyl ester through the use of concentrated acid in DCM provided the key intermediate tripeptidic acid (212) in 68% over 3 steps. The tripeptide 212 was then coupled with cyclopropyl amide amine 213 using EDCI, HOBt and N-methyl morpholine (NMM) to provide penultimate intermediate alcohol 234 in 95% yield. Subjection of -
Arzneimittelwechselwirkung
Potentially hazardous interactions with other drugs Alpha-blockers: avoid with alfuzosin.
Analgesics: risk of ventricular arrhythmias with methadone.
Anti-arrhythmics: risk of ventricular arrhythmias with amiodarone and disopyramide - avoid; risk of ventricular arrhythmias with flecainide andpropafenone - use with caution; use IV lidocaine with caution.
Antibacterials: concentration of both drugs increased with clarithromycin, erythromycin and telithromycin, increased risk of ventricular arrhythmias; avoid with rifabutin and rifampicin (concentration significantly reduced by rifampicin).
Anticoagulants: concentration of warfarin possibly affected; avoid with apixaban; possibly increased dabigatran concentration.
Antidepressants: possibly increased trazodone concentration; avoid with St John’s wort.
Antiepileptics: avoid with carbamazepine, fosphenytoin, phenobarbital, phenytoin and primidone.
Antifungals: concentration of both drugs possibly increased with ketoconazole, increased risk of ventricular arrhythmias; possibly increased itraconazole concentration; possibly increased posaconazole concentration - increased risk of ventricular arrhythmias; possibly altered voriconazole concentration - increased risk of ventricular arrhythmias.
Antipsychotics: avoid with pimozide; possibly increases lurasidone and quetiapine concentration - avoid.
Antivirals: concentration possibly reduced by atazanavir; concentration of atazanavir possibly increased; avoid with darunavir, fosamprenavir and lopinavir; concentration of daclatasvir and possibly olaparib increased - reduce daclatasvir and olaparib dose; concentration reduced by efavirenz - increase telaprevir dose; concentration possibly reduced by ritonavir; concentration of tenofovir possibly increased.
Anxiolytics and hypnotics: possibly increased midazolam concentration - risk of prolonged sedation, avoid concomitant use with oral midazolam.
Beta-blockers: risk of ventricular arrhythmias with sotalol - avoid.
Ciclosporin: concentration of both drugs increased, reduce ciclosporin dose.
Cilostazol: possibly increases cilostazol concentration.
Colchicine: possibly increased risk of colchicine toxicity - suspend or reduce colchicine dose, avoid in hepatic or renal impairment.
Cytotoxics: possibly increases bosutinib concentration - avoid or consider reducing dose of bosutinib; reduce dose of ruxolitinib.
Domperidone: possibly increased risk of ventricular arrhythmias - avoid.
Ergot alkaloids: avoid concomitant use.
Guanfacine: possibly increases guanfacine dose - halve dose of guanfacine.
Lipid-regulating drugs: avoid with lomitapide, simvastatin and atorvastatin.
Oestrogens: possibly reduced ethinylestradiol concentration and contraceptive effect.
Sildenafil: avoid concomitant use.
Sirolimus: concentration of both drugs increased, reduce sirolimus dose.
Beta2 sympathomimetics: avoid with salmeterol - risk of ventricular arrhythmias.
Tacrolimus: concentration of both drugs increased, reduce tacrolimus dose.
Tadalafil: avoid with high dose tadalafil.
Vardenafil: avoid concomitant use. -
Stoffwechsel
Extensively metabolised in the liver, involving hydrolysis, oxidation, and reduction.
Multiple metabolites were detected in faeces, plasma and urine. -
Lager
Store at -20°C
Telaprevir Anbieter Lieferant Produzent Hersteller Vertrieb Händler.
Global(293)Suppliers
-
Hebei Zhuanglai Chemical Trading Co Ltd
Telefon +86-16264648883<br/>+86-16264648883
E-Mail niki@zlchemi.com
-
Telefon +86-(0)57185586718<br/>+86-13336195806
E-Mail sales@capot.com
-
Beijing Cooperate Pharmaceutical Co.,Ltd
Telefon 010-60279497
E-Mail sales01@cooperate-pharm.com
-
Henan Tianfu Chemical Co.,Ltd.
Telefon +86-0371-55170693<br/>+86-19937530512
E-Mail info@tianfuchem.com
-
Telefon 008657128800458;<br/>+8615858145714
E-Mail FandaChem@Gmail.com
-
Shanghai Yingrui Biopharma Co., Ltd.
Telefon +86-21-33585366 - 03@
E-Mail sales03@shyrchem.com
-
Telefon +undefined-21-51877795
E-Mail ivan@atkchemical.com
-
Telefon +86-0371-86658258<br/>+8613203830695
E-Mail sales@coreychem.com
-
Telefon 0086-13720134139
E-Mail candy@biochempartner.com
-
Hubei xin bonus chemical co. LTD
Telefon 86-13657291602
E-Mail linda@hubeijusheng.com
1of2