坦索罗辛的用途及制备
发布日期:2019/5/21 9:19:26
概述[1]
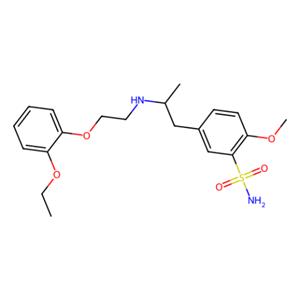
盐酸坦索罗辛(tamsulosinhydrochloride)是第三代超选择性长效α1的抑制剂,由日本山之内制药研发成功,1992年7月获FDA批准上市,商品名Harnal(哈乐),此后与勃林格殷格翰、雅培公司共同销售盐酸坦索罗辛,于1997年获FDA批准,商品名Flomax。该药能特异地抑制前列腺平滑肌的收缩,迅速缓解良性前列腺增生症临床症状,疗效好,不良反应更少,上市后销售额快速增长,是目前国内外畅销品种。
用途[2]
本品为一种对下尿道平滑肌具有强力且持续松弛作用的新型α1受体阻滞剂。而尿道、膀胱颈部及前列腺存在的α1受体主要为α1A受体,因此本品对尿道、膀胱颈部及前列腺平滑肌具有高选择性的阻断作用。适用于由前列腺增生引起的排尿困难、夜间尿频及残尿感等。
药物相互作用[3]
1.本品与其它肾上腺能阻滞剂合用可能影响其药代和药效动力学,建议二者不要合用。
2.西米替丁增加本品吸收并减少本品清除。合用时慎重,尤其本品剂量超过0.4mg时。
药理毒理[3]
本品属治疗良性前列腺增生症(BPH)用药,为选择性α1肾上腺素受体阻断剂。其主要作用机理是选择性地阻断前列腺中的α1A肾上腺素受体,松弛前列腺平滑肌,从而改善良性前列腺增生所致的排尿困难等症状。
药代动力学[3]
吸收、分布、消除:本品成人一次口服0.2mg时,6.8小时后血药浓度达到高峰,半衰期为10.0小时,其AUC0-∞与普通制剂几乎相等,因此是生物利用度没有降低的缓释制剂。连续口服,血药浓度可在第4天达到稳定状态。
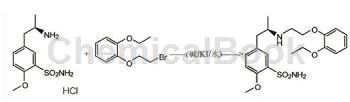
制备[1,4]
方法1:一种盐酸坦索罗辛的合成方法,所述的方法按以下步骤进行:
(1)缩合反应:在1000ml反应瓶中,加入RMBS(39.3g,98%,物质的量为0.245mol),EPEB0.245mol,氢氧化钾0.3675mol和乙腈,其中乙腈的用量为RMBS和EPEB总质量的3倍,升温至40℃,反应15h,然后降温、过滤,滤液减压蒸馏回收乙腈,得缩合物中间体53.2g,缩合物中间体的纯度94.8%,收率为93.5%(以RMBS质量为基准),缩合物中间体无须纯化直接进行下一步氢化反应;
(2)氢化反应:取缩合物中间体50g,在1000ml不锈钢氢化反应釜中加入缩合物中间体质量3倍的乙醇(150g)作为溶剂,加入含钯5%的钯碳催化剂2.5g,分别用氮气和氢气置换3次,再缓慢升温到30℃,通氢反应的压力0.5Mpa,反应10h后降温,滤去催化剂,滤液减压回收乙醇,所得固体用200ml纯化水打浆,洗涤,过滤,滤饼经真空干燥得类白色坦索罗辛游离碱38.6g,纯度为96.3%,收率为98.0%,重复步骤(1)和步骤(2)获得足量的坦索罗辛游离碱备用;
(3)成盐反应:取坦索罗辛游离碱142.0g,加入乙醇568g,加到2000ml反应瓶中,搅拌升温至30℃溶解,缓慢滴加工业盐酸,至反应液pH=2.0,反应6h后降温,抽滤,滤饼用乙醇洗涤,干燥得盐酸坦索罗辛124.7g,纯度99.5%,收率为83.3%(以坦索罗辛游离碱计)。
方法2:将R-(-)-5-(2-氨基丙基)-2-甲氧基苯磺酰胺盐酸盐(56g;0.2mol)、邻乙氧基苯氧乙基溴(49g;0.2mol)、碳酸钾(56g;0.4mol)加入到600ml水中,在85℃下,保温反应8小时,反应完毕后,冷至室温,抽滤,滤饼用100ml水洗涤、100ml甲醇洗涤,干燥得到56.6g的R-坦索罗辛游离碱,将56gR-坦索罗辛游离碱加入到500ml无水甲醇中,加热溶解,缓慢降温至25℃,搅拌析晶10小时,过滤,100ml无水甲醇洗涤,50℃鼓风干燥后得到50.8g的R-坦索罗辛游离碱(HPLC纯度99.91%,光学纯度(手性HPLC):S-坦索罗辛:0.032%)。
主要参考资料
[1] CN201310461289.2一种盐酸坦索罗辛的合成方法
[2] 临床实用药物手册
[3] 盐酸坦索罗辛缓释胶囊说明书
[4] CN201610894545.0制备稳定的盐酸坦索罗辛的方法


欢迎您浏览更多关于坦索罗辛的相关新闻资讯信息