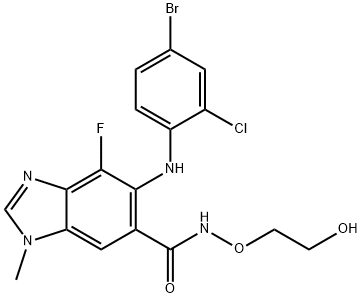
Selumetinib
- Product NameSelumetinib
- CAS606143-52-6
- MFC17H15BrClFN4O3
- MW457.68
- EINECS207-313-3
- MOL File606143-52-6.mol
Chemical Properties
| Melting point | >219°C (dec.) |
| Density | 1.69 |
| storage temp. | -20° |
| solubility | Soluble in DMSO (up to 50 mg/ml) or in Ethanol (up to 2 mg/ml) |
| form | Beige powder. |
| pka | 14.20±0.10(Predicted) |
| color | White |
| Stability | Stable for 1 year from date of purchase as supplied. Solutions in DMSO or ethanol may be stored at -20° for up to 2 months. |
| InChI | InChI=1S/C17H15BrClFN4O3/c1-24-8-21-16-13(24)7-10(17(26)23-27-5-4-25)15(14(16)20)22-12-3-2-9(18)6-11(12)19/h2-3,6-8,22,25H,4-5H2,1H3,(H,23,26) |
| InChIKey | CYOHGALHFOKKQC-UHFFFAOYSA-N |
| SMILES | C1N(C)C2=CC(C(NOCCO)=O)=C(NC3=CC=C(Br)C=C3Cl)C(F)=C2N=1 |
Selumetinib is an oral, potent selective mitogen activated protein kinase kinase (MEK) inhibitor, which has been shown to be effective against MEK-dependent tumours. It is intended to treat metastatic uveal melanoma, a rare malignancy that affects the eyes. It is the most common adult intraocular tumour; it arises from melanocytes in the uvea and affects mostly those from White ethnic groups, particularly those with light coloured irises.
Safety Information
| WGK Germany | WGK 2 |
| HS Code | 29349990 |
| Storage Class | 11 - Combustible Solids |
| Hazard Classifications | Acute Tox. 4 Oral Aquatic Chronic 4 Repr. 2 Skin Sens. 1 STOT RE 2 |
.")




