Researchers in France observed in 1953 that structures derived
from dehydrocholic acid, phenylethyl acetic acid, and certain
other disubstituted acetic acids exhibited hypocholesterolemic
properties in rats and humans. Several years later, Thorp and
Waring discovered clof ibrate as an effective compound for
lowering lipids in animal models, with minimal toxicity. Its
mode of action was initially attributed to seasonal variations in
adrenal and thyroid function, and the administration of
androsterone was found to potentiate the hypocholesterolemic
effect of this compound. Subsequently, several clinical trials
were performed which showed that clofibrate decreases lipid
levels in hypercholesterolemic patients, mainly as the result of
a reduction in the very-low-density lipoprotein (VLDL), and
less in the low-density lipoprotein (LDL) fraction, and that the
coadministration of androsterone was not necessary for its
hypolipidemic effect. Despite reported hepatomegaly in rats
following long-term treatment with clofibrate, this drug was
approved in the United States in 1967 for the treatment of
hyperlipidemias.
Clofibrate can be chemically synthesized by the condensation
of phenol with ethyl 2-chloro-2-methylpropionate in the
presence of a dehydrochlorinating agent, followed by chlorination
and purification. It can also be synthesized by the
condensation of p-chlorophenol with acetone and chloroform
followed by esterifying the resultant acid to give clofibrate.
Clofibrate is a selective agonist of peroxisome proliferator-activated receptor α (PPARα). In a transactivation assay, clofibrate exhibits EC50 values of 50 and 55 μM for murine and human PPARα, respectively. It also binds to PPARγ but with 10-fold less affinity and is inactive at PPARδ at concentrations up to 100 μM. Formulations containing clofibrate have been used to treat dyslipidemia and cardiovascular disease.
inhibits cholesterol biosynthesis
Clofibrate is a lipid-lowering agent (antilipidemic) used for
controlling high cholesterol (anticholesteremic) and triacylglyceride
levels in the blood. It increases lipoprotein lipase
activity to promote the conversion of VLDL to LDL, thereby reducing VLDL levels. It is indicated only in subjects with
increased concentrations of VLDL and intermediate-density
lipoproteins (IDL) who have failed to respond adequately to
gemfibrozil or nicotinic acid. Clofibrate is of limited utility for
patients with either familial hypercholesterolemia or polygenic
hypercholesterolemia, as comparatively more effective drugs
are available for lowering the concentration of LDL in these
patients.
Clofibrate has no effect on hyperchylomicronemia, nor
does it affect concentrations of high-density lipoproteins
(HDL). Thus, clofibrate appears to have specific efficacy only in
patients with familial type-III hyperlipoproteinemia. There is
no substantial evidence proving efficacy of clofibrate in preventing
deaths from coronary artery disease. Clofibrate has
been used to prevent or control polydipsia, polyuria, and
dehydration in a limited number of patients with mild to
moderate neurohypophyseal diabetes insipidus. A 5-year
multicenter study reported failure of clofibrate in reducing or
preventing mortality in cardiovascular disorders, which has
provided a setback for the prophylactic use of this drug.
ChEBI: The ethyl ester of clofibric acid.
The ethyl p-chlorophenoxyisobutyrate may be obtained by heating a mixture
of 206 parts of dry p-chlorophenoxyisobutyric acid, 1,000 parts of ethanol and
40 parts of concentrated sulfuric acid under reflux during 5 hours. The alcohol
is then distilled off and the residue is diluted with water and extracted with
chloroform. The chloroform extract is washed with sodium hydrogen carbonate
solution, dried over sodium sulfate and the chloroform removed by distillation.
The residue is distilled under reduced pressure and there is obtained ethyl p�chlorophenoxyisobutyrate, BP 148° to 150°C/20 mm.
The p-chlorophenoxyisobutyric acid used as starting material may be obtained
as follows. A mixture of 200 parts of p-chlorophenol, 1,000 parts of acetone
and 360 parts of sodium hydroxide pellets is heated under reflux and 240
parts of chloroform are gradually added at such a rate that the mixture
continues to reflux without further application of heat.
When addition is complete the mixture is heated under reflux during 5 hours
and then the acetone is removed by distillation. The residue is dissolved in
water, acidified with hydrochloric acid and the mixture extracted with
chloroform. The chloroform extract is stirred with sodium hydrogen carbonate
solution and the aqueous layer is separated. The alkaline extract is acidified
with hydrochloric acid and filtered. The solid product is drained free from oil
on a filter pump, then washed with petroleum ether (BP 40° to 60°C), and
dried at 50°C. The solid residue, MP 114° to 116°C, may be crystallized from
methanol (with the addition of charcoal) to give p-chlorophenoxyisobutyric
acid, MP 118° to 119°C.
Atromid (Wyeth);Aatroayerst;Aitiflus;Angiocapsul;Arterioflexion;Artriosan;Asa/cpib;Aterioplexin;Ateroayrest;Ateroclar;Aterofront;Ateronlen;Aterosol;Atevil;Atheroayerst;Atrofort;Atrolan;Atromid-s;Atrom-s;Ay 61;Biocleran;Clareden;Cloberab;Clobrate;Clobren-5 f;Clofenit;Clofibral;Clofibrate ayerst;Clofibrate compose;Clofibrato ayerst;Clofibrato procaps;Clofibrem;Clofimide;Clofin-icn;Clofipront 5000;Clofirem;Clofirin;Clofi-t;Clopin;Col 180;Contra-lipide;Corafen;Cr/085;Dabical;Dilectus;Doctus;Duplinal;Duraclofibrate;Ellemger;Eramid;Fibramid;Fibrolynt;Geri-70;Geromid;Healthstyle;Ici 28257nt;Ipolipid;Kontalipide;Levatram;Liapten;Liparil;Lipaten;Lipavlon 500;Lipicidon;Lipidicon;Liporan;Liptrinal;Lostat;Neoatromid;Nibratal;Nibratol;Nnormet;Nobret;Norinolipol;Normet richter;Nosterolin;Novofibrate;Omelip;Provasa;Recade;Regelan n 500;Sclerovasal;Serolipid;Sinteroid;Sklerocip;Sklerolip;Sklerovasal;Supraoxid;Tepincal;Tepingal;Vimedel;Vocaline.
World Health Organization (WHO)
Clofibrate, an antihyperlipidaemic agent, was introduced in 1967
and was subsequently extensively studied in the primary and secondary prevention
of ischaemic heart disease. Following reports, published in 1978, of increased
mortality among patients receiving clofibrate in a WHO-sponsored cooperative trial
concerned with the primary prevention of ischaemic heart disease, the drug was
withdrawn in some countries and its approved indications were severely restricted in many others. These restrictions have become the norm for more recently
developed analogues of clofibrate.
(Reference: (WHODI) WHO Drug Information, 2, 6, 1979)
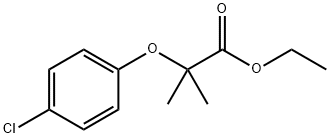
Clofibrate, ethyl 2-(p-chlorophenoxy)-2-methylpropionate (Atromid-S), is a stable, colorless topale yellow liquid with a faint odor and a characteristictaste. It is soluble in organic solvents but insoluble in water.
Clofibrate is prepared by a Williamson synthesis, condensingp-chlorophenol with ethyl -bromoisobutyrate, or bythe interaction of a mixture of acetone, p-chlorophenol, andchloroform in the presence of excess potassium hydroxide.The acid obtained by either of these methods is esterified togive clofibrate. Both acid and ester are active; the latter, however,is preferred for medicinal use. Clofibrate is hydrolyzedrapidly to 2-p-chlorophenoxy-2-methylpropionic acid by esterasesin vivo and, bound to serum albumin, circulates inblood. The acid has been investigated as a hypolipidemicagent. It is absorbed more slowly and to a smaller extent thanis the ester. The aluminum salt of the acid gives even lowerblood levels than p-chlorophenoxy-2-methylpropionic acid.
Questionable carcinogen; toxic; causes
nausea, vomiting, diarrhea, weakness, stiffness,
cramps, and muscle tenderness.
PPAR agonist (EC 50 values are 50, 500 and > 100 μ M at PPAR α , PPAR γ and PPAR δ respectively). Antihyperlipoproteinemic.
Clofibrate is a peroxisome proliferated activated receptor α (PPARα) agonist. It is a fibric acid derivative and has a therapeutic effect on hypertriglyceridemia and hyperlipoproteinemia type III. Clofibrate participates in lowering the very-low-density lipoprotein (VLDL) and cholesterol levels in hyperlipoproteinemia type III patients. It facilitates the decrease of total serum bilirubin concentration in Gilbert′s syndrome.
The three structurally related fibrates available in the
United States are gemfibrozil (Lopid), fenofibrate
(Tricor) and clofibrate (Atromid-S).They share common
uses and toxicities. The fibrates typically lower VLDL
triglyceride by 40% or more and elevate plasma HDL
cholesterol by 10 to 15%. The reduction of plasma
triglycerides in humans appears due to increased lipoprotein
lipase (LPL) activity. The fibrates activate a nuclear
receptor (transcription factor) termed peroxisomal proliferation
activated receptor (PPAR) that is a member of
the steroid hormone receptor superfamily. PPAR increases
transcription of the LPL gene and decreases transcription
of the apolipoprotein CIII gene (apo CIII).
Since LPL is responsible for catabolism of VLDL triglyceride
and apo CIII is an inhibitor of LPL activity, the
combined consequences of these changes are increased
LPL activity and enhanced removal of triglyceride from
the circulation.
The elevation of HDL levels by fibrates may be due
to two drug actions: induced synthesis of apo-A1, the
principal apoprotein of HDL, and increased assembly
of new HDL particles in the circulation. Surface components
of VLDL contribute to formation of HDL, as
the VLDL particles are reduced in size through the action
of LPL.The increased rate of catabolism of VLDL
caused by the fibrates would provide more components
for assembly of HDL particles.
The pro-drug, fenofibrate, requires a longer time to reach peak concentrations compared with gemfibrozil. Because of
differences in aromatic substitution, fenofibrate also has a much longer half-life than gemfibrozil. As previously mentioned, the
2,5-dimethyl substitution in gemfibrozil is much more susceptible to oxidative metabolism than the para-chloro group present in
fenofibrate. Similar to HMGRIs, changes in lipid levels are not seen immediately, and up to 2 months may be required to reach maximal
clinical effects and to determine the overall clinical efficacy.
Fibrates have excellent bioavailability and are extensively bound to plasma proteins. Because food can significantly enhance their oral
absorption, these compounds should be taken either with or just before meals. Fenofibrate was available in Europe and elsewhere as
standard tablet and capsule formulations for many years before its approval and marketing in the United States, where it was introduced
only after the development of a micronized formulation that allowed better oral absorption, a lower daily dose, and once-daily
administration. A 67-mg dose of micronized fenofibrate is bioequivalent to a 100-mg dose of nonmicronized drug. Since that time, two
additional tablet formulations have been developed. Abbott Laboratories currently markets TriCor as 48- and 145-mg tablets. The 48-mg
formulation is equivalent to previous 54- and 67-mg formulations, and the 145-mg tablet is equivalent to previous 160- and 200-mg
formulations. As noted in Table 30.10, fenofibrate is currently available in all of these strengths.
Renal elimination is the primary route through which these compounds are excreted from the body. Patients with mild renal dysfunction
often can be managed with minor dosage adjustments, whereas those with severe impairment or renal failure may have to discontinue its
use.
Clofibrate is the drug of choice in the treatment of typeIII hyperlipoproteinemias and may also be useful, to a lesserextent, in types IIb and IV hyperlipoproteinemias. The drugis not effective in types I and IIa.
Clofibrate can lower plasma concentrations of both triglyceridesand cholesterol, but it has a more consistent clinicaleffect on triglycerides. It also affects lipoprotein plasmalevels by enhancing removal of triglycerides from the circulationand causes reduction of VLDL by stimulatinglipoprotein lipase to increase the catabolism of this lipoproteinto LDL. Clofibrate lowers triglyceride levels in theserum much more than cholesterol levels and decreases levelsof FFAs and phospholipids. The lowering of cholesterollevels may result from more than one mechanism. Clofibrateinhibits the incorporation of acetate into the synthesis ofcholesterol, between the acetate and mevalonate step, by inhibitingsn-glyceryl-3-phosphate acyltransferase. Clofibratealso regulates cholesterol synthesis in the liver by inhibitingmicrosomal reduction of 3-hydroxy-3-methylglutaryl-CoA(HMG-CoA), catalyzed by HMG-CoA reductase. Clofibratemay lower plasma lipids by means other than impairment ofcholesterol biosynthesis, such as increasing excretionthrough the biliary tract.
The fibrates are generally well tolerated, with GI
distress being the most likely complaint. Other adverse
effects include myositis and erectile dysfunction, particularly
with clofibrate. There is ongoing concern about
the fibrates increasing the risk of gallstones, although
the extent of risk is unclear. Because clofibrate was associated
with increased mortality in early clinical trials,
it should be considered as a second-line drug.
Poison by intravenous route.Moderately toxic by ingestion and other routes. Anexperimental teratogen. Other experimental reproductiveeffects. Reduces plasma lipid levels. Human systemiceffects by ingestion: muscle weakness, muscle spasms, andfever. Q
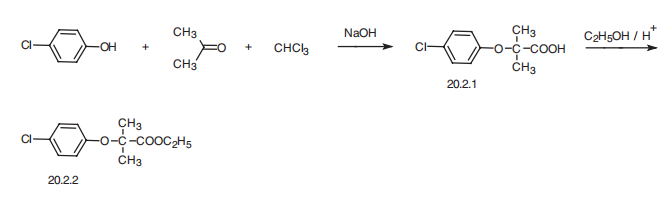
Clofibrate, ethyl ether 2-(4-chloropheoxy)-iso-butyric acid (20.2.2), is synthesized
by esterifying 2-(4-chlorophenoxy)-iso-butyric acid (20.2.1) with ethyl alcohol. This
is synthesized in a single-stage reaction from 4-chlorophenol, acetone, and chloroform in
the presence of an alkali, evidently by initial formation of chlorethone-trichloro-tert-butyl
alcohol, which under the reaction conditions is converted into (4-chlorophenoxy)trichlorotert-
butyl ether, and further hydrolyzed to the desired acid 20.2.1, which is further esterified
with ethanol in the presence of inorganic acid.
The fibrates potentiate the actions of the coumarin
anticoagulants, such as warfarin, so care should be taken
to reduce the dose of simultaneously administered anticoagulants,
and plasma prothrombin should be frequently
measured until the level stabilizes. As mentioned
earlier, great care should be given to combining
a statin with a fibrate, since this combination may increase
the risk of myositis and perhaps rhabdomyolysis.
Clofibrate characteristically reduces plasma triglycerides by
lowering the concentration of VLDL within 2–5 days after
initiation of therapy. In a majority of patients, total cholesterol
and LDL concentrations in plasma fall slightly. However, some
patients who exhibit a large fall in VLDL may show a paradoxical
rise in LDL, resulting in minimal net effect on total
cholesterol levels.
The drug has several proposed antilipidemic actions,
including increased triglyceride and VLDL clearance, mobilization
of cholesterol from tissues, increased fecal excretion of
neutral sterols, decreased hepatic lipoprotein synthesis and/or
secretion, decreased free fatty acid release, and decreased
triglyceride synthesis. The precise mechanisms by which clofibrate
lowers serum concentrations of triglycerides and cholesterol
are not known.
The pro-drug fenofibrate undergoes rapid hydrolysis to produce fenofibric acid. This active metabolite can then be further metabolized
by oxidative or conjugative pathways. Gemfibrozil is slightly different in that it does not require initial bioactivation; however, similar to
fenofibric acid, it can be oxidized or conjugated. Oxidation of the aromatic methyl groups produces inactive hydroxymethyl and carboxylic
acid analogues. As a drug class, fibrates and their oxidized analogues are primarily excreted as glucuronide conjugates in the urine.
Oxidization requires the CYP3A4 isozyme; however, because of the ability of these compounds to be conjugated and eliminated either
with or without oxidation, drug interactions with other compounds affecting the CYP3A4 system are less important here than with other
drug classes.
Clofibrate is a clear, colorless liquid with a density of 1.14 g ml-1
(at 25°C). The boiling point of clofibrate is 148–150°C at
25mmHg. This drug is a stable, colorless to pale-yellow liquid
with a faint odor and characteristic taste. Its melting point is
below 25°C, it is soluble in common solvents but not in water,
and its solubility or log P (octanol/water) is 3.620.
[1] Patent: WO2008/104994, 2008, A2. Location in patent: Page/Page column 32-33
[2] Journal of Medicinal Chemistry, 2017, vol. 60, # 2, p. 681 - 694
[3] Patent: WO2009/105509, 2009, A1. Location in patent: Page/Page column 36-37
[4] Bulletin de la Societe Chimique de France, 1956, p. 776,780, 781
[5] Patent: US2005/288329, 2005, A1. Location in patent: Page/Page column 44