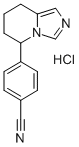
Fadrozole hydrochloride is a non-steroidal imidazole derivative launched for the treatment of post-menopausal breast cancer. Fadrozole is a potent and
specific aromatase inhibitor with neither androgenic nor estrogenic activities due to its
structural novelty. Fadrozole exerts its action by coordinating with the iron of the
porphyrin nucleus, presumably through the imidazole moiety. This strong binding
competes with the binding of molecular oxygen to iron and reversibly inactivates the
enzyme. An excellent clinical response rate has been reported for fadrozole with no
significant side effects.
5-p-Cyanophenyl-5,6,7,8-tetrahydroimidazo[1,5-a]pyridine hydrochloride:
A solution of 2.0 g of 4-(4-chloro-4-p-cyanophenyl-n-butyl)-1H-imidazole in
50 ml of chloroform is refluxed for 4 hours under nitrogen, cooled and
evaporated to yield the 5-p-cyanophenyl-5,6,7,8-tetrahydroimidazo[1,5-a]pyridine hydrochloride; melting point 231-233°C (from 2-propanol).
Preparation of the starting materials:
A solution of 1.82 g of 4-(3-ethoxycarbonylpropyl)-1H-imidazole in 30 ml of
tetrahydrofuran under nitrogen is treated with 0.5 g of sodium hydride (50%
oil dispersion) at 0°C for 30 min and 1.45 ml of trimethylsilyl chloride at 0°C
for 3 hours. The reaction mixture is washed with cold 0.5 N sodium
bicarbonate solution, dried over sodium sulfate and evaporated to dryness.
The oil is redissolved in 100 ml of methylene chloride at -78°C under nitrogen
and 12.82 ml of diisobutylaluminum hydride (1.56 M) is added dropwise. The
reaction mixture is stirred for 5 min at -78°C, quenched with 1 ml of
methanol followed by 10 ml of water and filtered through Celite. The organic
phase is separated, dried over sodium sulfate and evaporated to yield the title
compound (a).
(b) 4-(4-p-t-Butylaminocarbonylphenyl-4-hydroxy-n-butyl)-1-trimethylsilylim
idazole:6.95 g of p-tert-butylaminocarbonylbromobenzene is dissolved in 175 ml of
tetrahydrofuran at -70°C under nitrogen and 20.1 ml of a solution of n-butyl
lithium (2.7 m) in hexane is added dropwise. After reacting 30 min, a solution
of 5.69 g of 4-(3-formyl-n-propyl)-1-trimethylsilyl imidazole in 10 ml of
tetrahydrofuran is added slowly. The reaction mixture is allowed to warm
slowly to room temperature and 20 ml of ammonium chloride is added. The
organic layer is separated, dried over sodium sulfate and evaporated to yield
the title compound (b).
(c) 4-(4-Chloro-4-p-cyanophenyl-n-butyl)-1H-imidazole:
A solution of 4.5 g of 4-(4-p-t-butylaminocarbonylphenyl-4-hydroxy-n-butyl)-
1-trimethylsilylimidazole in 50 ml of thionyl chloride is refluxed for 1 hour,
cooled and evaporated. The residue is partitioned between methylene chloride
and aqueous sodium bicarbonate solution. The organic phase is separated,
dried over sodium sulfate and evaporated to yield the title compound (c).
5-p-Cyanophenyl-5,6,7,8-tetrahydroimidazo[1,5-a]pyridine:
A solution of 2.0 g of 4-(4-chloro-4-p-cyanophenyl-n-butyl)-1H-imidazole in
50 ml of chloroform is refluxed for 4 hours under nitrogen, cooled and
evaporated to yield the 5-p-cyanophenyl-5,6,7,8-tetrahydroimidazo[1,5-
a]pyridine.
Fadrozole is a nonsteroidal aromatase inhibitor. Fadrozole is a very potent and highly selective inhibitor of the aromatase enzyme system in vitro and estrogen biosynthesis in vivo. It inhibited the conversion of [4-14C]androstenedione to [4-14C]estrone by human placental microsomes in a competitive manner (Ki = 1.6 nM). At a substrate concentration 3-fold the Km, Fadrozole was 180 times more potent, as an inhibitor, than aminoglutethimide (Cat. No. A9657), exhibiting half-maximal inhibition at 1.7 nM as compared to 0.3 μM. In vivo, Fadrozole lowered ovarian estrogen synthesis by gonadotropin-primed, androstenedione treated, immature rats by 90% at a dose of 260 μg/kg (PO). In vivo, Fadrozole leads to sequelae of estrogen deprivation (e.g. regression of DMBA-induced mammary tumors) without causing adrenal hypertrophy in adult rats. It blocked aromatase by 50% in human breast cancer homogenates, live breast cancer cells, human placental microsomes, and porcine ovarian microsomes at concentrations of 0.008 to 0.02 μM.