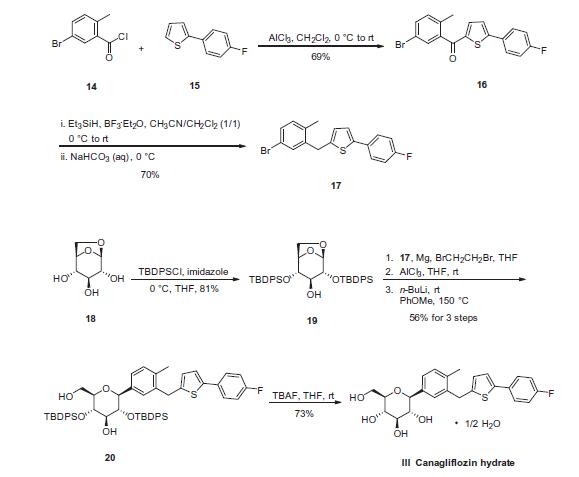
Canagliflozin heMihydrate
- Product NameCanagliflozin heMihydrate
- CAS928672-86-0
- MF2(C24H25FO5S).H2O
- MW462.53
- EINECS202-303-5
- MOL File928672-86-0.mol
Chemical Properties
| Melting point | 94-96°C |
| storage temp. | Hygroscopic, Refrigerator, under inert atmosphere |
| solubility | DMSO (Slightly), Methanol (Slightly) |
| form | Solid |
| color | White to Off-White |
| optical activity | 19.1°(C=0.01g/mL, MEOH, 20°C, 589nm) |
| Stability | Hygroscopic |
| InChIKey | RCCZPUWDQVUJAB-HNJBBDRUNA-N |
| SMILES | O[C@@H]1[C@H]([C@H](O)[C@@H](CO)O[C@H]1C1C=CC(C)=C(CC2=CC=C(C3C=CC(F)=CC=3)S2)C=1)O.O |&1:1,2,3,5,9,r| |