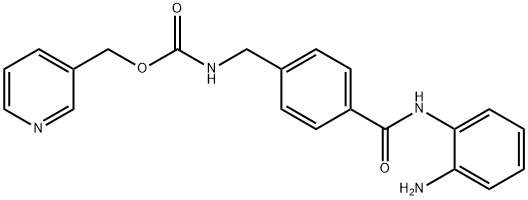
Entinostat
- Product NameEntinostat
- CAS209783-80-2
- MFC21H20N4O3
- MW376.41
- EINECS
- MOL File209783-80-2.mol
Chemical Properties
| Melting point | 159-160 ºC |
| Boiling point | 566.7±50.0 °C(Predicted) |
| Density | 1.315 |
| storage temp. | Keep in dark place,Sealed in dry,Store in freezer, under -20°C |
| solubility | DMSO: 38 mg/mL, soluble |
| form | solid |
| pka | 11.32±0.46(Predicted) |
| color | Pale yellow |
| Stability | Stable for 1 year from date of purchase as supplied. Solutions in DMSO may be stored at -20° for up to 1 week. |
| InChI | InChI=1S/C21H20N4O3/c22-18-5-1-2-6-19(18)25-20(26)17-9-7-15(8-10-17)13-24-21(27)28-14-16-4-3-11-23-12-16/h1-12H,13-14,22H2,(H,24,27)(H,25,26) |
| InChIKey | INVTYAOGFAGBOE-UHFFFAOYSA-N |
| SMILES | C(OCC1=CC=CN=C1)(=O)NCC1=CC=C(C(NC2=CC=CC=C2N)=O)C=C1 |
Safety Information
| Hazard Codes | Xi,T |
| Risk Statements | 36/37/38-62-48/25-25-61 |
| Safety Statements | 26-36-45 |
| RIDADR | UN 2811 6.1 / PGIII |
| WGK Germany | 3 |
| HS Code | 29333990 |
| Storage Class | 6.1C - Combustible acute toxic Cat.3 toxic compounds or compounds which causing chronic effects |
| Hazard Classifications | Acute Tox. 3 Oral Repr. 1A |
![4-[(PYRIDIN-3-YLMETHOXYCARBONYLAMINO)-METHYL]-BENZOIC ACID](/CAS/GIF/241809-79-0.gif)


