一种盐酸头孢唑兰的制备方法
发布日期:2022/1/4 14:03:33
背景技术
头孢唑兰(Cefozopran),化学名 1-[ [ (6R,7R)-7-[ [ (2Z) - (5-氨基 -1,2,4-噻二唑-3-基)(甲氧亚氨基)乙酰基]氨基]-2-羧酸-8-氧代-5-硫杂-1-氮杂双环[4. 2. 0]羊-2-烯-3-基]甲基]咪唑[1,2-b]咕嗪内盐。
头孢唑兰稳定性较差,因此在制备和使用时都是采用盐酸头孢唑兰(Cefozopranhydrochloride)形式。
盐酸头孢唑兰为日本武田药品工业公司研发的第四代注射用头孢菌素。头孢唑兰对黄色葡萄球菌的青霉索蛋白1、蛋白2及大肠菌及绿脓菌的青霉索蛋白3有高度亲和力,通过强效阻断细胞壁peptidoglycan架桥作用而阻碍细菌细胞壁的生成。可通过G-性菌外膜孔道迅速扩散到细菌周质并维持高浓度。具较低的β_内酰胺酶亲和性与诱导性,对染色体介导的和部分质粒介导的内酰胺酶稳定。因而对G+菌、G-菌、厌氧菌显示广谱抗菌活性,与第三代头孢菌素相比,增强了抗G+菌活性,特别对链球菌、肺炎球菌等有很强活性。头孢唑兰对一般头孢菌素不敏感的粪链球菌、弗劳地枸橼酸杆菌、阴沟肠杆菌、绿脓杆菌亦有较强作用。
根据文献报道,头孢唑兰主要有以下三种合成路线:
路线⑴:以7 β-氨基-3-(3-氧代丁酰氧基甲基)-3-头孢烯-4-羧酸(7-AACA)为原料,在五氯化磷和N,O-双三甲基硅基乙酰胺作用下与(Z)-2-(5-氨基-I,2,4-噻二唑-3-基)-2_甲氧亚氨基乙酸缩合,再在碘化钾作用下与咪唑并[1,2-b]哒嗪进行3-位取代,盐酸成盐得盐酸头孢唑兰,总收率为11.6%
路线(2) :7_AACA先用二碳酸二叔丁酯保护7位氨基,然后在碘化钠作用下与咪唑并[1,2-b]哒嗪进行3-位取代,经三氟乙酸脱保护,再在N-羟基苯并三唑和二环己基碳二亚胺作用下与(Z) -2- (5-氨基-1,2,4-噻二唑-3-基)-2-甲氧亚氨基乙酸缩合,盐酸成盐得盐酸头孢唑兰,总收率为9. 5%。
路线(3) :7_氨基头孢烷酸(7-ACA)经甲乙酸酐保护7_位氨基,在碘化钠作用下与咪唑并[1,2-b]哒嗪进行3-位取代,再经稀盐酸脱保护,然后与(Z)-2-(5-氨基_1,2,4-噻二唑-3-基)-2-甲氧亚氨基硫代乙酸(S-2-苯并噻唑)酯缩合,盐酸成盐得盐酸头孢唑兰,总收率为12.9%。
以7-ACA为中间体制备的头孢菌素品种中,有60 %以上的品种都可以用GCLE来生广。GCLE是氯甲基头抱菌素的商品名,化学名为7-苯乙酸胺基-3-氯甲基-3-头抱烯-4-羧酸对甲氧苯甲酯,其结构式如式(6)所示。其特点是3位上的氯增加了反应活性,而7位氨基、4位羧基均已被保护,在3位反应时可减少保护步骤。这一母核可替代7-ACA用于制造头孢菌素,尤其是对首先进行3位修饰的头孢菌素非常便利。而且,以GCLE为中间体生产头孢菌素时,产品收率更高、生产工艺更简单、生产条件更温和、产品成本更低,特别是在第四代头孢的合成上比7-ACA有非常大的优势。以GCLE为中间体生产头孢菌素是目前头孢合成的主要发展趋势。
在这些已知的合成方法中,采用7-ACA或7-AACA为原料制备头孢唑兰存在得率低,质量欠佳,且反应时间长,不利于连续性、大规模工业化生产。因此研究和开发工艺简单,高得率,高质量,并适合于大规模工业化生产的合成技术,已成为当务之急。
发明内容
本发明的目的在于针对现有技术的不足,提供一种采用GCLE制备高纯度、高收率盐酸头孢唑兰的方法,该方法简单易行、适用于产业化。
本发明的上述目的是通过如下方案予以实现的:
本发明提供一种盐酸头孢唑兰的制备方法,其特征在于包括如下步骤:(I)式(6)的GCLE与咪唑[1,2-b]并哒嗪反应生成式(5)化合物;
(2)式(5)化合物脱除C-4位的保护基形成式(4)化合物;
(3)式(4)化合物水解脱去7位的苯乙酰基得到式(3)的7-ACP ;
(4)式(3)的7-ACP与式⑵的头孢唑兰活性酯反应,制得式(1)的头孢唑兰,
反应方程式如下:
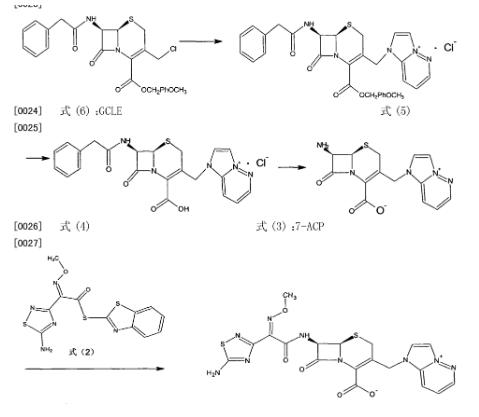
式⑴
上述所述制备方法,其特征在于所述步骤⑴中,GCLE与咪唑[1,2-b]并哒嗪在反应溶剂中反应,优选反应溶剂选自甲醇、乙醇、丙酮、乙腈、异丙醇中的一种或一种以上的混合溶剂,更优选为丙酮或乙腈。
上述所述制备方法,其特征在于所述步骤⑴中,GCLE与咪唑[1,2-b]并哒嗪反应温度为10〜50°c,优选为20〜40°C ;反应时间1〜10h,优选3〜7h。
上述所述制备方法,其特征在于所述步骤(1)中,将GCLE与咪唑[1,2-b]并哒嗪反应产物的溶液中,加入沉淀剂得到反应产物的沉淀,所述沉淀剂可选自水、甲醇、乙醇、异丙醇中的一种或一种以上的混合溶剂,优选为水、异丙醇中的一种或它们的混合溶剂。
上述所述制备方法,其特征在于所述步骤(2)中,式(5)化合物在脱羧剂作用下脱除C-4位的保护基形成羧基,所述的脱羧剂选自三氟乙酸、苯酚、间甲苯酚中的一种或一种以上的混合溶剂,优选为苯酚。
上述所述制备方法,其特征在于所述步骤(3)中,式⑷化合物酶水解脱去7位的苯乙酰基得到7-ACP,所述的酶优选为固定化青霉素酰化酶,更优选为PGA-450或IPA-750。
上述所述制备方法,其特征在于所述步骤(3)中,在弱碱性水溶液中,式(4)化合物酶水解脱去7位的苯乙酰基得到7-ACP,优选水溶液pH为7〜9,更优选为7. 3〜8。
上述所述制备方法,其特征在于所述步骤(3)中,7-ACP经过水洗去酶、消色洗涤后调解PH使得7-ACP沉淀析出;优选采用盐酸调解pH为1〜5,更优选pH为1. 5〜3,选pH为2〜2. 5。
上述所述制备方法,其特征在于所述步骤(4)中,7-ACP与头孢唑兰活性酯在反应溶剂中反应得到头孢唑兰,然后盐酸化得到盐酸头孢唑兰,所述反应溶剂可选自水、甲醇、乙醇、乙腈、乙酸乙酯、异丙醇、丙酮、三乙胺、二氯甲烷、三氯甲烷、正己烷、乙醚中的一种或一种以上的混合溶剂,优选水、甲醇、乙醇丙酮、三乙胺、二氯甲烷、三氯甲烷中的一种或一种以上的混合溶剂。
上述所述制备方法,其特征在于在易色变的步骤中进行消色处理,所述的消色处理可采用活性碳或还原剂以消除产物显色现象,所述还原剂可选自硫代硫酸盐,亚硫酸盐,亚硫酸氢盐、焦亚硫酸钠等中的一种或一种以上的混合。
作为优选,本发明所述的盐酸头孢唑兰生产方法,GCLE与咪唑[1,2-b]并哒嗪在反应溶剂中反应,然后在苯酚作用下,脱除C-4位的保护基形成羧基,再在水溶液中用酶水解脱去Ί位的苯乙酰基得到7-ACP,然后调节pH值使7-ACP析出。7-ACP与头孢唑兰活性酯(3)反应制备盐酸头孢唑兰。
具体实施方式
下面结合具体实施例对本发明做进一步地描述,但具体实施例并不对本发明做任何限定。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式等同落于本申请所附权利要求书所限定的范围。
(1)GCLE12. 5g,NaI4. 1g,丙酮80ml在30°C,搅拌反应15分钟。然后加入咪唑并哒嗪4. 5g,升温至35°C,搅拌5小时。加入异丙醇125ml,加毕搅拌30分钟。降温至0°C,搅拌1小时,过滤,滤饼用50ml异丙醇和30ml丙酮混合液洗涤两次。真空干燥,得黄绿色固体(GMPE)。加入苯酚25ml,升温至50°C,搅拌10小时。降温至40°C,加入10ml异丙醚和5ml异丙醇混合液,然后搅拌20分钟。将反应液加入到60ml异丙醚中,析出黄色固体,搅拌60分钟后过滤,滤饼用30ml丙酮和40ml异丙醚混合洗漆。滤饼加入到1. 2g的碳酸氢钠,45ml水,10ml丙酮的混合液中搅拌溶解。加入10ml异丙醚,搅拌15分钟,静置分层,下层加入0. 5g活性炭,脱色后过滤。滤液用2N盐酸或者6%碳酸氢钠溶液调节PH至8,升温至35℃。加入酶IPA-7501. 5g,水解5小时。过滤后加入0. 5g活性炭,O. 25g焦亚硫酸钠钠脱色20分钟,然后用5ml水洗涤。滤液用6N盐酸调节pH为2,然后降温0°C,搅拌60分钟后过滤,滤饼用冰水20ml洗涤,冰甲醇20ml洗涤。真空干燥至水分小于5%,得7-ACP。
(2) 7-ACP溶解在90ml甲醇中,降温至5°C,加入13ml三乙胺和10ml甲醇的混合溶液,搅拌溶解。然后加入头孢唑兰活性酯13. 5g,然后加入二氯甲烷25ml,搅拌反应8小时,高效液相色谱检测7-ACP小于2%。降温至5°C,搅拌I小时后,静置沉淀过滤得淡黄至类白色固体。加入64ml甲醇、32ml丙酮搅拌溶解后加入浓盐酸约2. 0ml,搅拌至溶解澄清,得淡黄绿色澄清溶液。加入活性碳0. 4g,脱色后过滤,用8ml甲醇和8ml丙酮混合溶液洗涤。将滤液转中快速加入300ml丙酮和300ml乙酸乙酯混合溶液,析出类白色固体,过滤抽干,得白色固体,真空干燥至水分小于3. 5%。得盐酸头孢唑兰约9. 3g,两步反应总收率65% (以 GCLE 计)。


欢迎您浏览更多关于头孢唑兰的相关新闻资讯信息