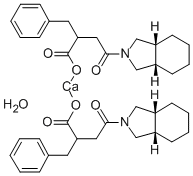
Description
Mitiglinide is the calcium salt form of Mitiglinide, which is a drug for the treatment of type II diabetes. It belongs to the meglitinide class blood glucose-lowering drug. Its mechanism of action is through stimulating the insulin secretion through closing the ATP-sensitive potassium channels in the pancreatic beta-cells. This process leads to depolarization, further stimulating the influx of calcium through the voltage-gated calcium channels. The high intracellular calcium level results in the exocytosis of insulin granules, alleviating the symptoms of type II diabetes.
Description
Mitiglinide is a non-sulfonylurea hypoglycemic agent that has been developed and
launched in Japan for the treatment of type-2 diabetes. Similar to the sulfonylurea
insulinotropic drugs, mitiglinide adopts a U-shaped configuration in which the base
of the U contains an amide linkage, and each branch of the U incorporates a
hydrophobic side chain. This similarity in conformation suggests that mitiglinide
also binds to the sulfonylurea receptor to cause the direct closing of ATP-sensitive
potassium channels in pancreatic β-cells; the result is stimulation of insulin secretion.
In contrast to typical sulfonylurea agents that frequently cause hypoglycemia
due to slowly reversed insulinotropic activity, mitiglinide’s short duration of action
should be advantageous in preventing this adverse effect. It also enjoys a rapid
onset of insulin release. Mitiglinide can be prepared by several closely related
methods, which involve either classical resolution of racemic intermediates, or enantioselective
methods, such as, chiral enolate alkylation, and asymmetric hydrogenation
with a rhodium or ruthenium-based chiral diphosphine complex. A highly
efficient preparative method for mitiglinide involves the diasteroselective alkylation
of a chiral acylsultam intermediate that is obtained by the reaction of 3-phenylpropionyl
chloride with (-)-camphorsultam. The chiral enolate of the acylsultam is
derived by using sodium hexamethyldisilazane as the base, and is subsequently alkylated
with tert-butyl bromoacetate to achieve >93% diastereomeric purity of the
product. Following cleavage of the tert-butyl ester with trifluoroacetic acid, the resultant
acid is coupled with (3aR,7aS)octahydro-1H-isoindole, and the camphorsultam
chiral auxiliary is removed by saponification to produce the parent acid of
mitiglinide in high yield. In vitro, mitiglinide has about a 1000-fold greater affinity for
the Kir6.2/SUR1 form of potassium-ATP channels expressed in β-cells (IC
50=4nM)
than for the Kir6.2/SUR2A or Kir6.2/SUR2B channel types found in cardiac and
smooth muscle. In fact, it is significantly less potent in blocking potassium-ATP
channels than the prototype sulfonylurea glyburide (IC
50=42μM vs. 0.13 μM, respectively);
thus, it possesses a more favorable cardiac safety profile. Phase III clinical
data demonstrated that mitiglinide significantly improved indices of blood glucose
control (postprandial glucose and fasting plasma glucose levels) in a double blind,
comparative study. It was also confirmed that the incidence of hypoglycemia, a frequent
adverse effect, remained as low as placebo. In another placebo-controlled
study involving twenty-two patients with type-2 diabetes, mitiglinide 5mg t.i.d.
treatment significantly suppressed postprandial plasma glucose elevations (181 vs.
261mg/dL with placebo), and the daily change in blood glucose level was reduced
with no subjective symptoms. No episodes of hypoglycemia or abnormal clinical
laboratory parameters were noted. Regarding the pharmacokinetics, a single oral
dose (unspecified) of mitiglinide reached its peak plasma concentration of
about 0.5 μg/mL at 30 minutes post dose and then steadily declined to about
0.04 μg/mL at 4 h.
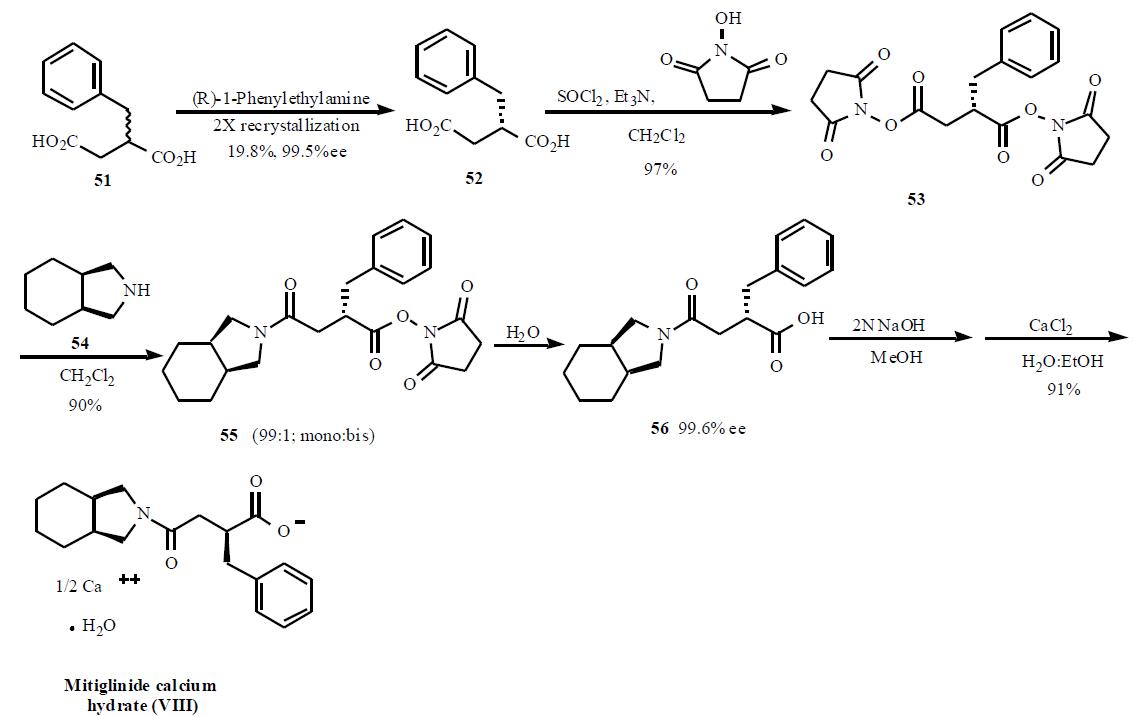
Synthesis
A number of publications and
patents have disclosed the syntheses of mitiglinide.
One of the syntheses describing the preparation of
mitiglinide using bis-activated esters to obtain a selective
mono amide is described in Scheme 8. The synthesis starts
with racemic 2-benzylsuccinic acid (51) which was resolved into its enantiomer using chiral amine salt formation and
crystallization. Out of several amines used, (R)-1-
phenylethylamine gave the best results for the chiral
resolution (99.5% ee, 19.5%). Acid 52 was treated with
thionyl chloride and triethylamine followed by Nhydroxysuccinamide
to give doubly activated ester 53
(97%). Treatment of this double ester 53 with
tetrahydroisoindoline (54) gave selectively mono amide
to di-amide in 99:1 ratio. Hydrolysis of the activated ester in
55 with water gave desired product 56 in 99% yield.
Subsequent conversion in two steps to the half calcium salt
provided mitiglinide calcium hydrate (VIII) in 91% yield.
Mode of action
Mitiglinide is another non-sulfonylurea insulin secretagogue. Insulin secretion is physiologically stimulated by the binding of ATP to a cytosolic nucleotide binding site of the membrane bound ATP-sensitive K+ channel which leads to closure of the K+ channel. The inhibition of K+ permeability depolarizes the plasma membrane, subsequently the voltage-dependent Ca2+ channel opens to promote the Ca2+ influx which finally results in insulin secretion. The insulin secretagogues stimulate insulin secretion by closure of this ATP-sensitive K+ channel of pancreatic -cells. KATP-channel consists of two subunits, a functional ion channel pore and a regulatory protein. The binding of the secretagogues is suggested to occur to the separate regulator protein containing the binding sites for sulfonylureas (sulfonylurea receptor 1, SUR1) but also other compounds.